Note
Go to the end to download the full example code.
Analysing and clustering cohort sequences#
Scenario: Starting from the cohort prepared in Filtering and preparing a cohort, you want to quantify how similar the admission sequences are across patients, group them into clusters with shared patterns, and visualise the results.
Concepts covered:
Compute a pairwise sequence distance matrix with
HammingEntityMetric+LinearPairwiseSequenceMetricCluster with
HierarchicalClustererInspect cluster membership
Produce a faceted timeline coloured by cluster
Imports#
import polars as pl
from tanat.clustering import HierarchicalClusterer
from tanat.criterion import (
EntityCriterion,
LengthCriterion,
)
from tanat.dataset import access
from tanat.metric import HammingEntityMetric, LinearPairwiseSequenceMetric
from tanat.sequence.type.interval.pool import IntervalSequencePool
from tanat.visualization import SequenceVisualizer
Rebuild the prepared cohort#
Self-contained rebuild using the builder API (see Exploring a patient cohort and Filtering and preparing a cohort for details).
DB = f"sqlite:///{access('mimic4')}"
pool = IntervalSequencePool(
store=(
IntervalSequencePool.builder()
.add_sql(
DB,
"SELECT subject_id, admittime, dischtime,"
" admission_type, admission_location"
' FROM "hosp/admissions"',
id_column="subject_id",
start_column="admittime",
end_column="dischtime",
features=["admission_type", "admission_location"],
)
.add_sql(
DB,
'SELECT subject_id, gender, anchor_age AS age FROM "hosp/patients"',
id_column="subject_id",
is_static=True,
features=["gender", "age"],
)
.build("admissions_store", exist_ok=True)
)
)
# ``pl.Categorical`` is required by the metric and clustering modules, and
# enables consistent colour-coding across all visualisations.
pool.cast_features({"admission_type": pl.Categorical}, is_static=False)
┌─ Interval SequenceStore
│
│ Step 1/4: Sorting & preparing data
│
│ Step 2/4: Building sequence index
│
│ Step 3/4: Writing entity, time index & static features
│
│ Step 4/4: Computing & writing metadata
│
└─ Done (100 sequences · 275 entities · 0.01s)
print(pool)
┌────────────────────────────────────────────────┐
│ IntervalSequencePool Summary │
└────────────────────────────────────────────────┘
Overview
─────────────────────────
Sequences 100
Store /home/runner/.tanat_workspace/building_pools_tutorial/admissions_store
id_column id
Time Index
─────────────────────────
Type Datetime(time_unit='us', time_zone=None) [2110-04-11 15:08:00 → 2201-12-17 13:45:00]
Columns ['start', 'end']
t0 position=0, anchor=start
Entity Features (2)
─────────────────────────
• admission_location String [len 4 → 38]
• admission_type Categorical (9 categories)
Static Features (2)
─────────────────────────
• age String [len 2 → 2]
• gender String [len 1 → 1]
Cohort selection#
Same selection as Filtering and preparing a cohort: patients with at least 2 admissions who experienced at least one emergency, aligned to their first emergency (T0).
ids_cohort = pool.which(LengthCriterion(ge=2)) & pool.which(
EntityCriterion(query=pl.col("admission_type") == "EW EMER.")
)
print(f"[Intersection] → {len(ids_cohort)} IDs")
[which] LengthCriterion → 48 / 100 IDs (48.0%)
[which] EntityCriterion → 60 / 100 IDs (60.0%)
[Intersection] → 36 IDs
cohort = pool.subset(ids_cohort)
# Align sequences to first emergency admission (T0).
cohort.set_t0(query=pl.col("admission_type") == "EW EMER.", anchor="start")
print(cohort)
┌────────────────────────────────────────────────┐
│ IntervalSequencePool Summary │
└────────────────────────────────────────────────┘
Overview
─────────────────────────
Sequences 36
Store /home/runner/.tanat_workspace/building_pools_tutorial/admissions_store
id_column id
Time Index
─────────────────────────
Type Datetime(time_unit='us', time_zone=None) [2114-03-19 20:05:00 → 2201-12-17 13:45:00]
Columns ['start', 'end']
t0 query, anchor=start
Entity Features (2)
─────────────────────────
• admission_location String [len 4 → 38]
• admission_type Categorical (9 categories)
Static Features (2)
─────────────────────────
• age String [len 2 → 2]
• gender String [len 1 → 1]
Step 1: Define the sequence metric#
HammingEntityMetric compares two admissions at the same
position: distance 0 if they share the same type, 1 otherwise.
LinearPairwiseSequenceMetric aggregates these
entity-level distances along the full sequence. The padding_penalty
penalises sequences of different lengths.
entity_metric = HammingEntityMetric(entity_feature="admission_type")
sequence_metric = LinearPairwiseSequenceMetric(
entity_metric=entity_metric,
padding_penalty=1.0,
)
Step 2: Compute the distance matrix#
dist_matrix = sequence_metric.compute_matrix(cohort)
┌─ LinearPairwiseSequenceMetric
│
│ Chunks: 0%| | 0/1 [00:00<?, ?it/s]
│ Chunks: 100%|██████████| 1/1 [00:00<00:00, 3100.00it/s]
│
└─ Done (36 sequences · 0.00s)
dm = dist_matrix.data
print(f"Distance matrix shape : {dist_matrix.shape}")
Distance matrix shape : (36, 36)
Step 3: Hierarchical clustering#
We group patients into 3 clusters using complete-linkage hierarchical
clustering. After fit(),
the cluster label is automatically added as a static feature on the pool
under the name given by cluster_column.
clusterer = HierarchicalClusterer(
metric=sequence_metric,
n_clusters=3,
linkage="complete",
cluster_column="adm_cluster",
)
clusterer.fit(cohort)
┌─ HierarchicalClusterer
│
│ Step 1/2: Computing distance matrix
│
│ ┌─ LinearPairwiseSequenceMetric
│ │
│ │ Chunks: 0%| | 0/1 [00:00<?, ?it/s]
│ │ Chunks: 100%|██████████| 1/1 [00:00<00:00, 5753.50it/s]
│ │
│ └─ Done (36 sequences · 0.00s)
│
│ Step 2/2: Clustering (HierarchicalClusterer)
│
└─ Done (36 items, 3 clusters · 0.02s)
HierarchicalClusterer(clusters=3)
Step 4: Inspect cluster membership#
clusters exposes the fitted
Cluster objects directly. Each provides a
size and the list of patient items. The cluster label is also
available as a static feature (adm_cluster) for downstream filtering
or visualisation.
for cluster in clusterer.clusters:
print(cluster)
Cluster(id=0, size=19)
Cluster(id=1, size=7)
Cluster(id=2, size=10)
Cluster labels are also stored as a static feature for downstream use.
cohort.static_data().head()
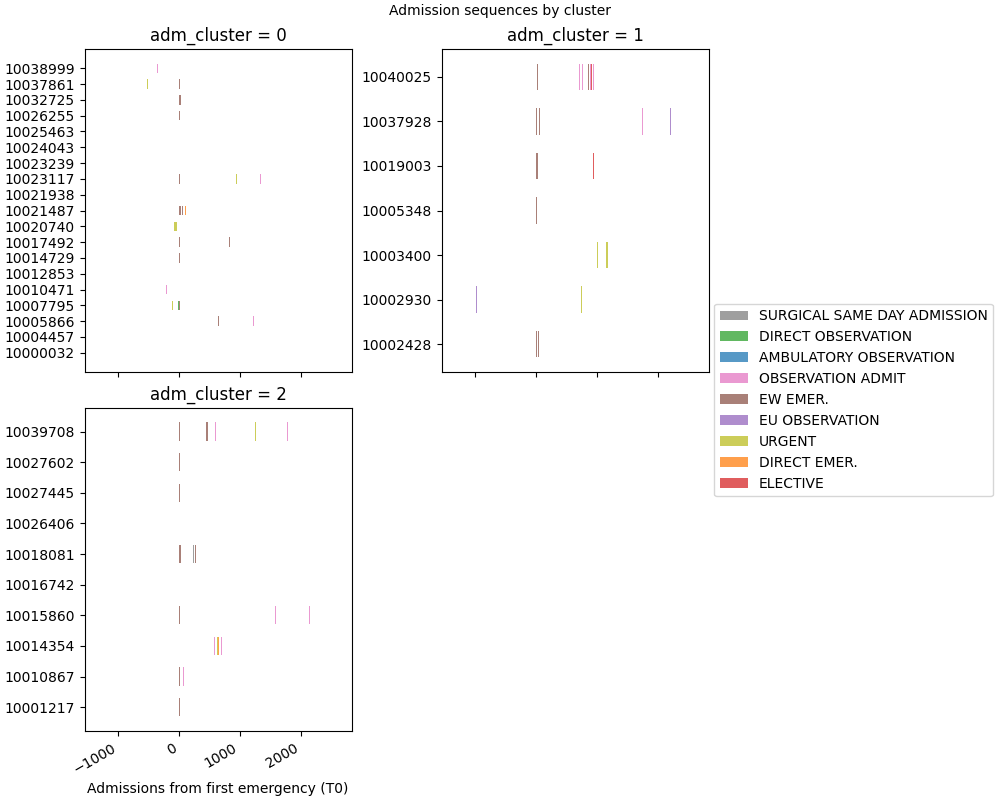
Step 5: Faceted timeline coloured by cluster#
Each panel shows the admission sequences of one cluster, aligned to T0. This makes it easy to spot structural differences between groups.
fmt: off
SequenceVisualizer.timeline(time_mode="relative", allow_large=True) \
.title("Admission sequences by cluster") \
.x_axis(label="Admissions from first emergency (T0)") \
.colors("tab10") \
.facet(by="adm_cluster", is_static=True, cols=2, share_y=False) \
.draw(cohort, entity_feature="admission_type") \
.show()
# fmt: on
Total running time of the script: (0 minutes 0.969 seconds)